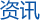
本标准规定了纤维级聚酯切片各分析项目所使用的试剂,仪器,试验步骤和允许误差。
本标准适用于对纤维级聚酯切片的仲裁检验。
2引用标准
GB 8170数值修约规则
3试验方法
3.1特性粘度的测试
3.1.1概述
在25℃测定溶剂和浓度为0.005 g/mL的聚酯溶液的流出时间。由这些测定值和已知溶液的浓度计算特性粘度。在本方法中动能校正系数是很小的,可不予考虑。
3.1.2溶剂
苯酚-四氯乙烷(化学纯)混合液(质量比1:1),使用前均需分别蒸馏,二种溶剂以质量比1:1混合,可在干燥箱中于60℃下加热混合物,不时摇动,直到完全均一状态,经标定(比重法、折光法、紫外光谱法等均可使用),确认配比正确后贮于棕色瓶中。
3.1.3仪器
3.1.3.1恒温水浴:控制水浴温度25±0.05℃。
3.1.3.2乌氏粘度计:粘度计尺寸见下图。对溶剂和使用温度下的流经时间应大于200 s,半微量粘度当粘度计毛细管直径为0.58~1.03 mm时,C球容积为4.0 mL(±5%),P管的内径为6.0 mm(±5%)。

3.1.3.3容量瓶:25 mI。
3.1.3.4分析天平:感量0.000 1 g。
3.1.3.5耐酸砂芯漏斗:5-2#。
3.1.3.6加热板或蒸汽浴。
3.1.3.7真空干燥箱:压力小于133.3 Pa,温度为100±2℃。
3.1.4试验步骤
3.1.4.1 测定溶剂流出时间
将约17 mL的苯酚一四氯乙烷溶液经砂芯漏斗过滤后,充入粘度计中,并将其置于25±0.05℃的水浴中,保持15~20 min后,测量苯酚-四氯乙烷溶液的流出时间,取5次的测定值(其误差不超过±0.05 s),求平均值作为纯溶剂的流出时间t0;
3.1.4.2试样的测定
在分析天平上精确称取已剪碎的试样0.125 0±0.000 1 g,放入25 mL容量瓶中。
如样品的含水量高于0.5%,则需将聚合物放在真空干燥箱(3.1.3.7)中干燥,压力低于133.3 Pa,温度为100℃,时间3 h。然后在干燥器中冷却称量。
在上述容量瓶中加入15~20 mL溶剂,置于加热板或蒸汽浴中加热,使样品全部溶解(溶解条件为90~100℃,约1 h),取出,冷却后置于25±0.05℃的水浴槽中,恒温20 min,用保持在浴槽温度的溶剂加到容量瓶内之溶液体积到刻线,并充分混合。
将上述溶液经砂芯漏斗过滤后充入粘度计中,在恒温水浴中(25±0.05℃)放置20 min,然后测其流出时间,重复测量三次,相互间误差不得大于0.1 s,求出平均值作为溶液流出时间t0。
3.1.5结果计算
计算到小数点第四位,根据数值修约规则取到小数点后第三位。

3.2软化点的测试
3.2.1仪器和工具
3.2.1.1软化点测定装置。
3.2.1.2热风循环干燥器。
3.2.1.3锉。
3.2.1.4 刀片。
3.2.1.5试管:声16 mm×150 mm(平底)。
3.2.1.6试管架。
3.2.1.7氯乙烯板。
3.2.1.8硅油。
3.2.1.9接点复活剂(三合净化剂)。
3.2.2试验步骤
3.2.2.1 把试样切片和软化点标准切片各装入试管中。
3.2.2.2在140℃时热处理60 min。
3.2.2.3 冷却后用刀具使其成型,使角型切片成4 mm×4 mm×2 mm大小,圆柱形切片修成直径3 mm厚度2 mm大小,再修整成上下两平面无凸凹状。
3.2.2.4设定油浴上限温度为270℃,下限温度为240℃,以10℃/min的升温速率升至240℃。
3.2.2.5从油浴中提起软化点检测器的测微计。
3.2.2.6用接点复活剂除掉可动接点部分套管间的灰尘。
3.2.2.7把试样和标准切片安置在检测器的温度计上,把柱塞放在切片中央,一个周期放二个标准切片,其余放试样,一个试样测两点,记录标准切片和试样的柱塞编号。
3.2.2.8旋转测微计,使其下降到和可动接点部分接触,再使测微计的刻度和零重合(柱塞表示灯亮)。
3.2.2.9反向旋转测微计和可动接点距离为0.5 mm。
3.2.2.10再次确认柱塞在各片样品中央,然后把检测器放入已升温至240℃的油浴中。
3.2.2.11 用900~1 200 r/min的转速和1.0~1.2℃/min的升温速度,搅拌、加热油浴。
3.2.2.12当某柱塞灯亮时,表示此样测试结束,即记下测量温度,当所有的柱塞灯全亮后,表示标准切片和各试样的测试均已结束,可切断加热器、搅拌马达及记录仪。
3.2.3结果计算

3.2.4同一样品二点测定值之差不应超过0.3℃。
3.3熔点的测试
3.3.1仪器
3.3.1.1样品切片机。
3.3.1.2偏光显微镜。
3.3.1.3升温控制单元(包括加热台,控制装置,释放器)。
3.3.1.4载玻片:厚度1mm。
3.3.1.5盖玻片:18 mm×18 mm×0.17 mm。
3.3.2试剂
熔点标准物:225℃,237℃,247℃,260℃。
3.3.3温度指示的校正
3.3.3.1 每次系列试验前,用熔点标准物校正温度指示。
3.3.3.2取适量的熔点标准物放于载玻片上,用盖玻片压紧晶粒,使其互相接触,在显微镜下观察是一个单层。
3.3.3.3将装有标准物的载玻片放在加热台上,以2℃/min的速率升温。
3.3.3.4在显微镜下观察,当晶粒引起的光效应消失时,即按下释放器上的按钮A,所显示的温度即为该标准物的熔点。
3.3.3.5根据标准物的熔点和显示出的温度,计算出温度指示的校正值。
3.3.4试验步骤
3.3.4.1 用样品切片机将切片切成厚为25 μm的薄片,再用剪刀剪取约0.5 mm2的样品,放在载玻片上,用盖玻片压紧。
3.3.4.2将载玻片放在加热台上,快速升温至180℃,然后以10℃/min的速率升温,至240℃后再以2℃/min的升温速率升温,观察初熔,记下读数。
3.3.4.3样品达到初熔后,快速升温至280℃,使其在此温度下保持3 min,然后快速降低到180℃,再以10℃/min的升温速率升温至240℃,最后以2℃/min的速率升温,观察终熔,现象同3.3.3.4,所显示的温度即为样品的熔点。
3.3.5同一样品的二次测定值之差不应超过0.5℃。
3.4羧基的测试
3.4.1仪器
3.4.1.1 自动电位滴定仪,包括滴定架,玻璃电极和甘汞电极。
3.4.1.2加热板(可恒温调节)。
3.4.1.3锥形瓶:300 mL,标准磨口。
3.4.1.4 回流冷凝器:标准磨口。
3.4.1.5气体洗瓶。
3.4.1.6干燥塔(装有干燥剂)。
3.4.1.7磁力搅拌器。
3.4.1.8五通联接头的滴定装置(二个用于电极,二个用于氮气出、入,一个接滴定管)。
3.4.1.9分析天平:感量0.000 1 g。
3.4.2试剂
3.4.2.1邻甲酚:分析纯,需经蒸馏提纯(一周内使用)。
3.4.2.2氯仿:分析纯。
3.4.2.3乙醇或苯甲醇:分析纯(苯甲醇需经蒸馏提纯)。
3.4.2.4草酸:分析纯。
3.4.2.5氢氧化钾:分析纯。
3.4.3试验步骤
3.4.3.1 氢氧化钾-乙醇(或苯甲醇)溶液(约0.05 mol/L)的配制标定
准确称取2.85 g氢氧化钾溶于1 L乙醇或苯甲醇中。
精确称取20~30 mg草酸,准确至0.1 mg加入50 mL,经煮沸后冷却的蒸馏水,溶解后进行电位滴定。

称量后倒入300 mL锥形瓶中,准确加入50 mL7:3(质量比)的邻甲酚与氯仿混合液,装上回流冷凝器,在沸腾温度下回流30 min溶解试样,然后在室温下冷却。
3.4.3.3滴定:将溶解好的溶液定量地转移到滴定装置中,玻璃电极和甘汞电极依次用水、乙醇、氯仿冲洗后浸入溶液中,开启电磁搅拌器,并通入经干燥塔干燥过的氮气,5 min后,用0.05 mol/L氢氧化钾一乙醇溶液滴定,自动电位仪将自动记录滴定曲线,滴定曲线的拐点为滴定的等当点。
使用过的电极依次用氯仿、乙醇、水冲洗后留待使用。
3.4.3.4用相同条件做空白试验。
3.4.4结果计算
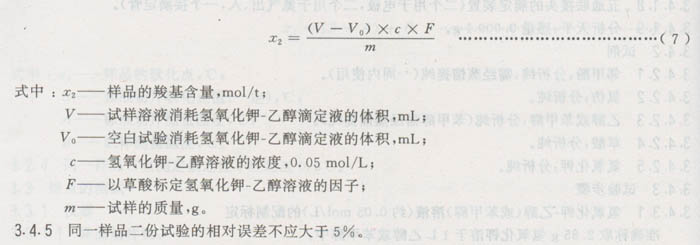
3.4.5同一样品二份试验的相对误差不应大于5%。
3.5色度的测试
A法
3.5.1 仪器
3.5.1.1 自动色差计,光源D65,10°视野,自动测定x、y、z系统的数值,并转换到L、a、b系统来表示色度数据。
3.5.1.2试样容器。
3.5.1.3标准板(每年计量校验一次)。
3.5.1.4测量用杯。
3.5.1.5玻璃皿。
3.5.1.6热风循环干燥器。
3.5.1.7样筛:20目、40目。
3.5.2试验步骤
3.5.2.1 开启仪器,视仪器说明书规定预热一定时间,将标准板放入载物台上,用粗调微调刻度盘,将x、y、z标准值调入。
3.5.2.2把试样放入干燥箱中,在135±5℃下加热30 min(有光切片加热60 min),使之结晶化。冷却后,粉碎过筛,取20~40目的颗粒。
3.5.2.3在测量用杯内放入筛得的样品颗粒,抖平,放于载物台上,测定试样的L值和b值。
3.5.2.4把试样从载物台上放回已加热结晶化后的样品中,再放入测量杯中重复测试二次,取同一样品的三次算术平均值。
三次测试中,当L值差异大于1.5,6值差异大于0.5时,则进行再次测定。
B法(非仲裁用)
除样品不需粉碎过筛外,其余和A法相同。
3.6凝集粒子的测试
3.6.1仪器和工具
3.6.1.1显微镜(目镜10倍,物镜25倍)及显微镜照明灯。
3.6.1.2样品切片机。
3.6.1.3分析天平:感量0.000 1 g。
3.6.1.4盖玻片:18 mm×18 mm×0.17 mm。
3.6.1.5镊子、医用剪刀。
3.6.1.6计数器。
3.6.1.7测微尺。
3.6.1.8载玻片。
3.6.2试剂
浸油N20D=1.51
3.6.3操作步骤
3.6.3.1样品制备
将0.5~1 kg切片样品,按四分法进行取舍,直至留下10 g切片样品,从切片样品中取5粒切片,并且用调整到20 μm的切片机将每粒切5片,取所切的片若干片,使之总量为5 mg,将这些切片放在洁净的载玻片上,并用浸油将其很好地覆盖,把盖玻片紧紧地压在样品上,使形成一个平整的表面。
3.6.3.2测定凝集粒子
把装有切片的载玻片放到显微镜台上,调节显微镜焦距,用透射光寻找大于和等于10 μm的凝集粒子(椭圆形的粒子算最长的部分)。
3.6.3.3计算结果
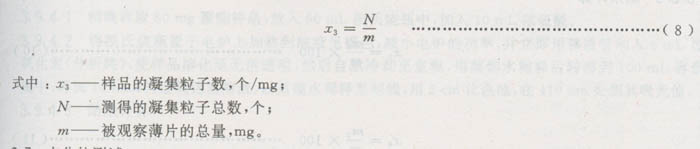
3.7水分的测试
3.7.1 仪器
3.7.1.1分析天平:感量0.000 1 g。
3.7.1.2真空干燥箱:使用范围20~200℃。
3.7.1.3称量瓶(或铝盒):Φ65 mm×35 mm。
3.7.1.4干燥器:Φ210 mm×320 mm。
3.7.2试验步骤
3.7.2.1把称量瓶(或铝盒)打开盖子放在真空干燥箱中,在箱中残余压力低于400 Pa,温度120℃的条件下干燥2 h。除去真空,打开干燥箱,把称量瓶瓶盖盖好后迅速移入干燥器中,冷却1 h后称量(准确到O.000 1 g)。
3.7.2.2在上述称量瓶中称入约20 g试样(准确到0.000 1 g)。按3.7.2.1步骤操作。
3.7.3结果计算
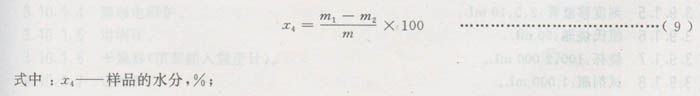

3.7.4同一样品两份试验的相对误差应不大于5%。
3.8粉末和异状切片的测试
3.8.1仪器和工具
3.8.1.1 取样器。
3.8.1.2分析天平:感量0.000 1 g。
3.8.1.3塑料盘。
3.8.1.4称量瓶:Φ70 mm×30 mm。
3.8.1.5毛刷。
3.8.1.6样筛:20目。
3.8.2试验步骤
3.8.2.1用取样器称取试样100 g。
3.8.2.2把样筛置放塑料盘上,然后将样品倒入样筛。
3.8.2.3用样筛筛试样,把粉末筛入塑料盘中。
3.8.2.4从留在样筛的切片中,检出长度长于、等于正常切片的4倍,厚度、宽度或直径大于或等于正常切片的2倍的粒子作异状切片。
3.8.2.5称取粉末和异状切片的量。
3.8.3结果计算

3.9二氧化钛含量的测试
3.9.1 仪器
3.9.1.1分光光度计:备有1、2、3、5 cm的比色池。
3.9.1.2分析天平:感量0.000 1 g。
3.9.1.3电炉:附调压变压器。
3.9.1.4容量瓶:100、1 000 mL。
3.9.1.5刻度移液管:2、5、10 mL。
3.9.1.6凯氏烧瓶:50 mL。
3.9.1.7烧杯:100、2 000 mL。
3.9.1.8试剂瓶:1 000 mL。
3.9.2试剂
3•9.2.1硫酸:分析纯(d=1.84)。
3.9.2.2硫酸铵:分析纯。
3.9.2.3硫酸溶液:5 mol/L。
3.9.2.4过氧化氢:分析纯。
3.9.2.5过氧化氢溶液:3%。
3.9.2.6二氧化钛:分析纯(纯度99.9%)。
3•9•2•7分解液:将700 mL浓硫酸加到2 000 mL烧杯中,置于电炉上加热至沸,立即加入400 g硫酸铵,加热溶解,然后自然冷却至室温,装入1 000 mL试剂瓶。
3.9.2.8钛标准溶液(1 mg/mL)。
将166.8 mg二氧化钛(相当于100 mg钛)和20 mL分解液加到100 mL烧杯中加热溶解,自然冷却后用蒸馏水稀释,将此溶液移至100 mL容量瓶中,用蒸馏水稀释至刻度线,摇匀。
3.9.3工作曲线的绘制
3•9•3.1用刻度移液管移取3.9.2.8钛标准溶液0、0.2、0.4、0.6、0.8、1.0、1.2、1.4、1.6 mL分别注入9只100 mL容量瓶中。
3•9.3.2在上述容量瓶中,各加入50 mL蒸馏水和20 mL硫酸溶液(5 mol/L),摇匀后用移液管各加入10 mL 3%过氧化氢溶液,最后以蒸馏水稀释至刻线,摇匀。
3•9•3.3在分光光度计上,以410 nm为工作波长,用5 cm比色池测定3.9.3.2各瓶中溶液的吸光值。
3.9.3.4根据测定的吸光值及对应的钛含量绘制工作曲线。
3.9.4试验步骤
3.9.4.1精确称取50 mg聚酯样品,放入50 mL凯氏烧瓶中,加入10 mL浓硫酸。
3•9.4.2将凯氏烧瓶置于电炉上加热到溶液呈褐色,减小电炉的功率,并立即用移液管加入5 mL过氧化氢(分析纯),使样品溶化至无色透明,然后自然冷却至室温,用蒸馏水稀释后转移到100 mL容量瓶中,再加10 mL3%过氧化氢溶液,以蒸馏水稀释至刻线,用5 cm比色池,在410 nm处测其吸光值。
3.9.4.3结果计算
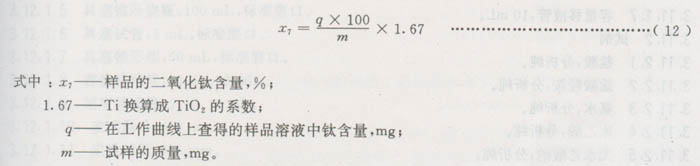
3.9.5同一样品的两份试验的相对误差应不大于2%。
3.10灰分的测试
3.10.1仪器
3.10.1.1分析天平:感量0.000 1 g。
3.10.1.2瓷坩埚:50 mL。
3.10.1.3 电炉。
3.10.1.4高温电阻炉。
3.10.1.5坩埚钳。
3.10.1.6干燥器(顶部插入温度计)。
3.10.1.7定时钟。
3.10.2试验步骤
3.10.2.1 把瓷坩埚放入高温电阻炉中,于850℃灼烧60 min,取出后移至干燥器中,冷却60 min,称得坩埚重量,准确至0.000 1 g。重复上述步骤,直到灼烧至恒重(两次称量之差不大于0.4 mg)。
3.10.2.2在已恒重的坩埚中称入适量样品(半消光切片称2 g,不含消光剂的切片称20 g),放在电炉上,缓慢地不燃烧地进行炭化,直至全部试样炭化完毕。
3.10.2.3把上述坩埚转移到400℃的高温电阻炉中灼烧至不冒烟,再将炉温升至850℃,继续灼烧60 min。取出后移至干燥器中,冷却60 min,称得残渣质量。
3.10.2.4结果计算
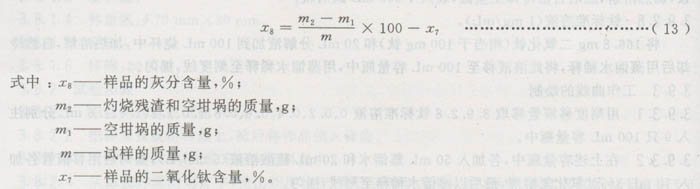
3.10.3同一样品的两份试验的相对误差应不大于4%。
3.11铁分的测试
3.11.1 仪器
3.11.1.1分光光度计:备有1、2、3、5 cm的比色池。
3.11.1.2分析天平:感量0.000 1 g。
3.11.1.3 电炉。
3.11.1.4容量瓶:100、500、1 000mL。
3.11.1.5刻度移液管:5 mL。
3.11.1.6量杯:20 mL。
3.11.1.7容量移液管:10 mL。
3.11.2试剂
3.11.2.1盐酸:分析纯。
3.11.2.2盐酸羟胺:分析纯。
3.11.2.3氨水:分析纯。
3.11.2.4冰乙酸:分析纯。
3.11.2.5无水乙酸钠:分析纯。
3.11.2.6硫酸铁铵[NH4Fe(SO4)2•12H2O]:分析纯。
3.11.2.7 pH值为4.5的乙酸-乙酸钠缓冲溶液
将164 g无水乙酸钠溶解在大约500 mL的水中,加240 mL冰乙酸,用蒸馏水稀释至1 000 mL。
3.11.2.8邻菲罗啉:分析纯。
3.11.2.9铁标准溶液(0.020 0 mg/mL)
称取硫酸铁铵1.727 g溶解在200 mL的水中,定量地移到1 000 mL容量瓶中,加20 mL硫酸溶液(9 mol/L),稀释至刻线,并混合。移取此溶液50 mL到500 mL的容量瓶中,稀释至刻线并混和。
本标准溶液也可用纯度为99.9%的铁粉来制备。
3.11.3工作曲线的绘制
3.11.3.1 用刻度移液管移取3.11.2.9铁标准溶液0、0.5、1.0、1.5、2.0、2.5、3.0 mL分别注入100 mL容量瓶中,各加入40 mL左右蒸馏水。
3.11.3.2各瓶中加入5 mL4%的盐酸羟铵溶液摇匀,放置数分钟后,以约为3 mol/L的氨水调节pH至4左右,再加入10 mL乙酸-乙酸钠溶液。待溶液冷却至室温后再加入5 mL浓度为1 g/L的邻菲罗啉溶液,用蒸馏水稀释至刻线,摇匀。
3.11.3.3在分光光度计上以510 nm为工作波长,5 cm比色池,测定3.11.3.2各瓶内溶液之吸光值。
3.11.3.4根据测得的吸光值及对应的铁含量绘制工作曲线。
3.11.4试验步骤
3.11-4.1称取10.0 g样品,放于瓷坩埚中按3.10.2.2至3.10.2.3要求进行灰化。在灰化残分里加入5 mL盐酸溶液(约5 mol/L),在电炉上加热加速铁分的溶解,取下后经滤纸滤入100 mL容量瓶中,以40 mL蒸馏水分多次反复冲洗滤纸及滤纸中残留物,滤液一并进入瓶中。
3.11.4.2按3.11.3.2~3.11.3.4步骤测得样品液的吸光值。
3.11.4.3结果计算
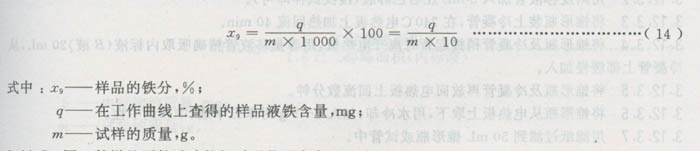
3.11.5同一份样品两份试验的相对误差不应大于5%。
3.12二甘醇的测试
3.12.1仪器
3.12.1.1气相色谱仪,带有氢火焰离子化检测器。
3.12.1.2微型注射器:10μL。
3.12.1.3电热板:200℃以上。
3.12.1.4球型冷凝管。
3.12.1.5具塞锥形烧瓶:100 mL,标准磨口。
3.12.1.6具塞试管:5 mL,标准磨口。
3.12.1.7具塞锥形瓶:50 mL,标准磨口。
3.12.1.8容量移液管:10、15、20 mL。
3.12.1.9刻度移液管:5、10、15 mL。
3.12.1.10玻璃漏斗:Φ50 mm。
3.12.1.11定性滤纸:Φ90 mm。
3.12.1.12分析天平:感量0.000 1 g。
3.12.1.13台天平。
3.12.1.14样筛:20目、40目。
3.12.1.15水浴锅。
3.12.1.16烧杯:400 mL。
3.12.1.17真空干燥箱。
3.12.2试剂
3.12.2.1 乙醇胺:分析纯。
3.12.2.2甲醇:分析纯。
3.12.2.3丙酮:分析纯。
3.12.2.4对苯二甲酸(TPA):工业品。
3.12.2.5内标液。
A液:16己二醇(1,6-Hexanedi01)色谱纯,5±0.05 g加100 mL甲醇。
B液:取A液10 mL,用甲醇稀释至2 000 mL(1,6己二醇5 mg/20 mL)。
C液:取A液5 mL,用甲醇稀释至100 mL(25 mg/10 mL)。
3.12.2.6二甘醇溶液
二甘醇(diethylene glyc01)色谱纯。
二甘醇0.25 g,加甲醇200 mL。
3.12.2.7气相色谱担体:白色硅藻土担体(20-40目)。
3.12.2.8气相色谱固定液聚乙二醇一分子量20 000(PEG-20M)。
3.12.3试验步骤
3.12.3.1称取试样1 g(准确到0.000 1 g),放入锥形瓶内。
3.12.3.2用刻度移液管加入3 mL左右乙醇胺(浸没试样即可)。
3.12.3.3将锥形瓶装上冷凝管,在240℃电热板上加热回流40 min。
3.12.3.4将锥形瓶及冷凝管稍提起暂时离开电热板,用容量移液管精确吸取内标液(B液)20 mL,从冷凝管上部缓慢加入。
3.12.3.5将锥形瓶及冷凝管再放回电热板上回流数分钟。
3.12.3.6将锥形瓶从电热板上取下,用水冷却,加入4~5 g对苯二甲酸,中和到中性。
3.12.3.7用滤纸过滤到50 mL锥形瓶或试管中。
3.12.3.8用微量注射器吸取1~2 μL清液注入气相色谱仪中进行测试。
3.12.4气相色谱仪测试条件
3.12.4.1氮气:25~35 mL/min。
3.12.4.2氢气:20~30 mL/min(检测器不点火时,严禁开启氢气针形阀以防止氢气泄入空间引起爆炸)。
3.12.4.3空气:700~900 mL/min。
3.12.4.4柱温:165~170℃。
3.12.4.5检测器温度:175~185℃。
3.12.4.6汽化室温度:200~210℃。
3.12.4.7注入量:1~2μL。
3.12.4.8色谱柱:PEG一20M5%/白色硅藻土担体(20~40目)玻璃柱Φ3.5 mm×2 250 mm。
3.12.4.9记录纸速度:10 mm/min。
3.12.5色谱柱制作方法
3.12.5.1称取必要量的担体。
3.12.5.2称取对担体质量比为5%的固定液,用对担体体积为80%的甲醇量进行溶解、摇匀(操作过程中要防止甲醇溅入眼睛)。
3.12.5.3将担体倒入盛有固定液的烧杯中,加入甲醇将担体完全浸没。
3.12.5.4在水浴(60℃)中用玻璃棒轻轻搅匀,并让甲醇慢慢地蒸发(甲醇蒸气对人体有害,须在排气橱内进行)。
3.12.5.5放入真空干燥箱中(60℃)干燥2 h。
3.12.5.6冷却后用20~40目样筛进行分离。
3.12.5.7将涂渍完毕的固定相充填入洗净并干燥好的色谱柱中(用真空泵边抽气边填入)。
3.12.5.8以氮气为载气,压力0.2 MPa,180℃下老化24 h。
3.12.6检量系数K的确定
3.12.6.1取5个100 mL的容量瓶,每只瓶中各加入内标液C液10 mL。
3.12.6.2分别逐个加入二甘醇标准溶液5、10、20、40、80 mL。
3.12.6.3以甲醇稀释到刻度线,摇匀。
3.12.6.4将各容量瓶的溶液注入气相色谱仪中(注入量同3.12.3.8)。
3.12.6.5求内标物的PA比及mt比,以5只数据的平均值作为K值。
3.12.7计算
聚酯切片中二甘醇含量:

附加说明:
本标准由中华人民共和国纺织工业部提出。
本标准由上海化学纤维公司归口。
本标准由上海化学纤维公司测试中心、燕山石化公司聚酯厂、仪征化学纤维工业联合公司、上海石化总厂联合起草。江苏省纺织工业厅、天津石化公司涤纶厂、扬州合成化工总厂协助起草。
本标准主要起草人瞿德方、阎淑焕、史维洵、夏蕴琴。
本标准参照采用国际标准ISO1628/1—1984塑料、聚合物稀溶液粘度和特性粘度测定方法的标准化准则。ISO6685工业用化工产品——铁含量测定的通用方法——邻菲哕啉分光光度法。ASTMD2857—70聚合物稀溶液粘度标准试验方法。JIS Z 8722—1982物体色的测定。BS 4603—1970化学产品灼烧或灰分残分的试验方法。